Analysis Results
Results from running the pipeline on the chr21 cohort: 22 samples (12 ALS, 10 CTRL), bisulfite-sequenced cfDNA.
Fragment length distribution
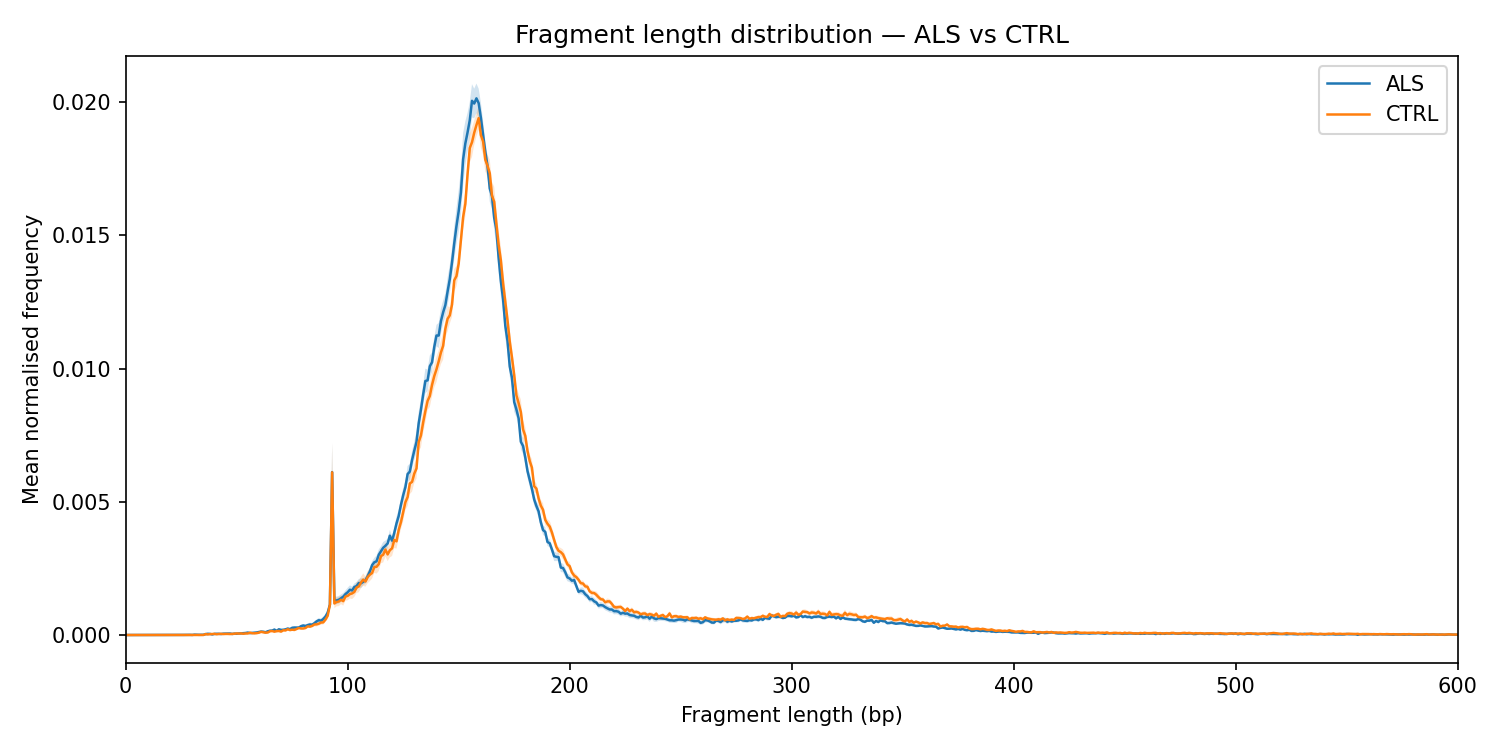
A mono-nucleosomal peak at ~167 bp is visible in both groups. ALS samples show a higher mono-nucleosomal / di-nucleosomal ratio (fl_ratio_mono_di), the strongest discriminating feature in this dataset (effect size 0.84).
Read position distributions
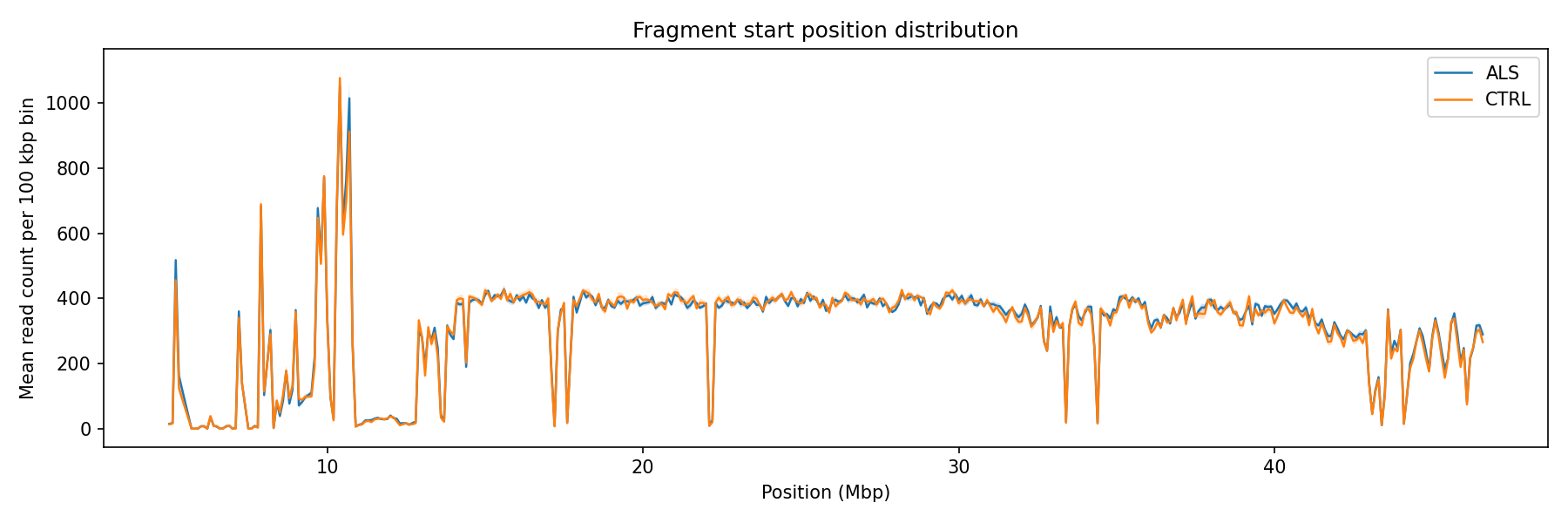
Distribution of fragment 5′ start and 3′ end positions across chr21 in 100 kbp bins. Peaks correspond to regions with higher read depth; troughs reflect low-mappability regions (centromere, pericentromeric repeats). The two groups track closely, with no strong regional enrichment distinguishing ALS from CTRL on chr21.
End-motif distribution
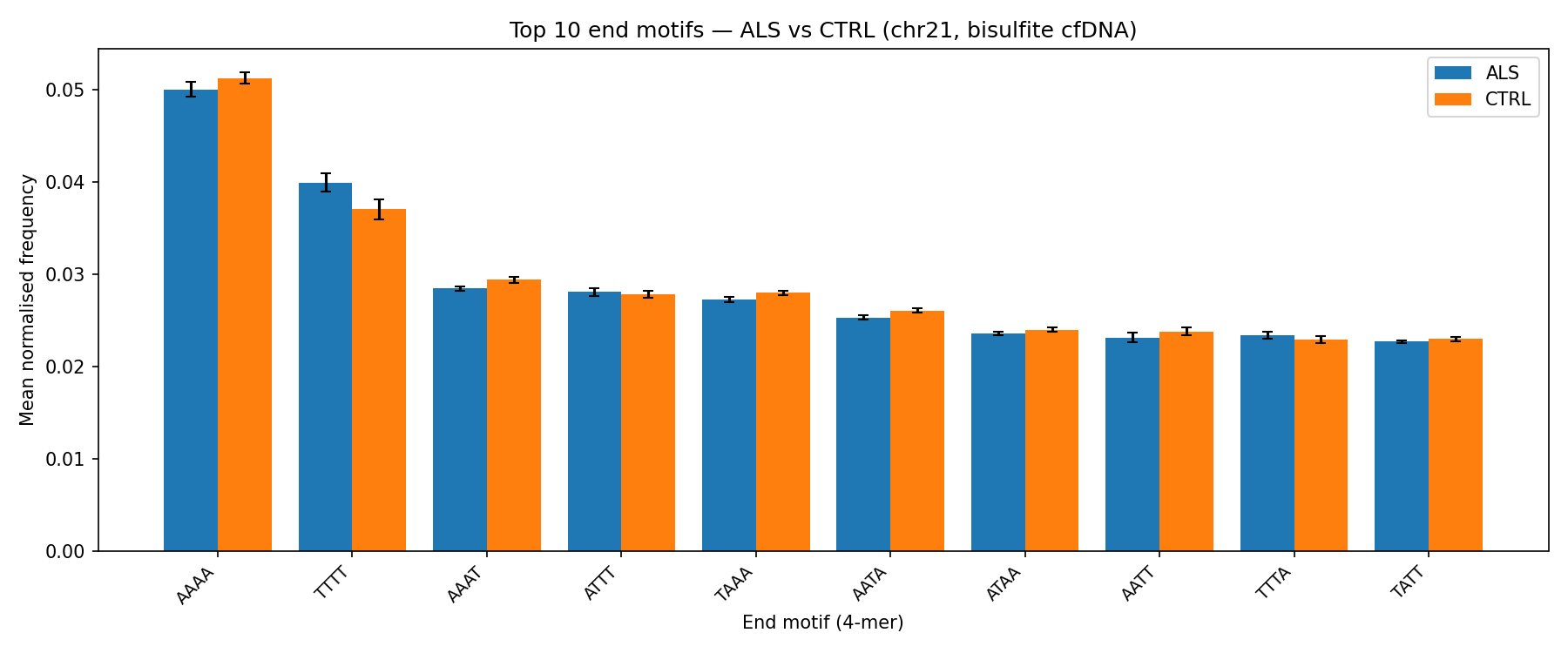
Frequencies of the top end 4-mers (5′ end of each cfDNA fragment). The end-motif spectrum reflects nuclease preference (DNase I, CAD). Bisulfite conversion biases the distribution toward T/A-rich motifs and compresses inter-sample variance.
Methylation summary
Coverage-weighted mean methylation per sample. chr21 is constitutively methylated across both groups so methylation features contribute negligible discriminative signal in this cohort.
Feature contributions
Running LOO-CV with methylation features excluded raises accuracy from 0.64 to 0.68 (macro F1 0.62 → 0.66). The per-feature effect sizes confirm the cause:
| Feature | ALS mean | CTRL mean | Effect size (Cohen's d) |
|---|---|---|---|
fl_ratio_mono_di |
16.3 | 12.7 | 0.84 |
fl_std |
57.2 | 64.2 | 0.57 |
fl_mean |
169.8 | 175.6 | 0.53 |
fl_frac_nucleosomal |
0.797 | 0.775 | 0.48 |
methylation_entropy |
0.0187 | 0.0187 | 0.01 |
methylation_median |
1.000 | 1.000 | 0.00 |
Effect size is Cohen's d: abs(mean_ALS - mean_CTRL) / pooled_sd, where pooled_sd = sqrt(((n1-1)*s1^2 + (n2-1)*s2^2) / (n1+n2-2)).
methylation_median = 1.0 for both groups: the majority of covered CpG sites on chr21 are fully methylated in every sample. chr21 is dominated by satellite repeats and constitutive heterochromatin that are uniformly methylated regardless of disease state. Including the methylation features adds five near-zero-signal dimensions to a 27-feature space with n=22, and the regulariser cannot fully suppress them.
Methylation features are retained in the code because they may carry genuine signal in full-genome runs (differentially methylated regions in promoters, enhancers, imprinted loci) and add little computational overhead at extraction time.
Classification
LOO-CV with L2 logistic regression (inner GridSearchCV for regularisation strength):
| All features | Fragment-only | |
|---|---|---|
| Accuracy | 0.64 | 0.68 |
| Macro F1 | 0.62 | 0.66 |
ALS sensitivity (recall) is 75% — the model identifies 9 of 12 ALS cases. Specificity (CTRL recall) is 50% — it correctly labels 5 of 10 controls. ALS precision is 64%: of samples called ALS, 64% are true positives. These metrics reflect the chr21 limitation: with no ALS-specific signal on chr21, the classifier is working largely from global cfDNA biology, and the regulariser is under-constrained at n=22.
Full classification report (all-features run):
precision recall f1-score support
als 0.64 0.75 0.69 12
ctrl 0.62 0.50 0.56 10
accuracy 0.64 22
macro avg 0.63 0.62 0.62 22
weighted avg 0.63 0.64 0.63 22
Fragment length features drive classification; methylation features add noise on chr21. See Classification Methodology for the full methodology and leakage-prevention details.
Chr21 scope
Chr21 is ~1.5% of the autosomal genome. Extending to the full genome is expected to increase classification power, particularly for methylation features which are uninformative here but may carry genuine signal at promoters, enhancers, and imprinted loci elsewhere.
Downloads
- sample_summary.csv — per-sample extracted features (fragment length, methylation statistics)